Simulated annealing
Simulated annealing (SA) is a generic probabilistic metaheuristic for the global optimization problem of applied mathematics, namely locating a good approximation to the global optimum of a given function in a large search space. It is often used when the search space is discrete (e.g., all tours that visit a given set of cities). For certain problems, simulated annealing may be more effective than exhaustive enumeration — provided that the goal is merely to find an acceptably good solution in a fixed amount of time, rather than the best possible solution.
The name and inspiration come from annealing in metallurgy, a technique involving heating and controlled cooling of a material to increase the size of its crystals and reduce their defects. The heat causes the atoms to become unstuck from their initial positions (a local minimum of the internal energy) and wander randomly through states of higher energy; the slow cooling gives them more chances of finding configurations with lower internal energy than the initial one.
By analogy with this physical process, each step of the SA algorithm replaces the current solution by a random "nearby" solution, chosen with a probability that depends on the difference between the corresponding function values and on a global parameter T (called the temperature), that is gradually decreased during the process. The dependency is such that the current solution changes almost randomly when T is large, but increasingly "downhill" as T goes to zero. The allowance for "uphill" moves saves the method from becoming stuck at local optima—which are the bane of greedier methods.
The method was independently described by Scott Kirkpatrick, C. Daniel Gelatt and Mario P. Vecchi in 1983,[1] and by Vlado Černý in 1985.[2] The method is an adaptation of the Metropolis-Hastings algorithm, a Monte Carlo method to generate sample states of a thermodynamic system, invented by N. Metropolis et al. in 1953.[3]
Contents |
Overview
In the simulated annealing (SA) method, each point s of the search space is analogous to a state of some physical system, and the function E(s) to be minimized is analogous to the internal energy of the system in that state. The goal is to bring the system, from an arbitrary initial state, to a state with the minimum possible energy.
The basic iteration
At each step, the SA heuristic considers some neighbour s' of the current state s, and probabilistically decides between moving the system to state s' or staying in state s. The probabilities are chosen so that the system ultimately tends to move to states of lower energy. Typically this step is repeated until the system reaches a state that is good enough for the application, or until a given computation budget has been exhausted.
The neighbours of a state
The neighbours of a state are new states of the problem that are produced after altering the given state in some particular way. For example, in the traveling salesman problem, each state is typically defined as a particular permutation of the cities to be visited. The neighbours of some particular permutation are the permutations that are produced for example by interchanging a pair of adjacent cities. The action taken to alter the solution in order to find neighbouring solutions is called "move" and different "moves" give different neighbours. These moves usually result in minimal alterations of the solution, as the previous example depicts, in order to help an algorithm to optimize the solution to the maximum extent and also to retain the already optimum parts of the solution and affect only the suboptimum parts. In the previous example, the parts of the solution are the parts of the tour.
Searching for neighbours to a state is fundamental to optimization because the final solution will come after a tour of successive neighbours. Simple heuristics move by finding best neighbour after best neighbour and stop when they have reached a solution which has no neighbours that are better solutions. The problem with this approach is that a solution that does not have any immediate neighbours that are better solution is not necessarily the optimum. It would be the optimum if it was shown that any kind of alteration of the solution does not give a better solution and not just a particular kind of alteration. For this reason it is said that simple heuristics can only reach local optima and not the global optimum. Metaheuristics, although they also optimize through the neighbourhood approach, differ from heuristics in that they can move through neighbours that are worse solutions than the current solution. Simulated Annealing in particular doesn't even try to find the best neighbour. The reason for this is that the search can no longer stop in a local optimum and in theory, if the metaheuristic can run for an infinite amount of time, the global optimum will be found.
Acceptance probabilities
The probability of making the transition from the current state  to a candidate new state
to a candidate new state 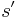 is specified by an acceptance probability function
is specified by an acceptance probability function 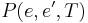 , that depends on the energies
, that depends on the energies 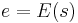 and
and 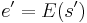 of the two states, and on a global time-varying parameter
of the two states, and on a global time-varying parameter  called the temperature.
called the temperature.
One essential requirement for the probability function  is that it must be nonzero when
is that it must be nonzero when 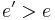 , meaning that the system may move to the new state even when it is worse (has a higher energy) than the current one. It is this feature that prevents the method from becoming stuck in a local minimum—a state that is worse than the global minimum, yet better than any of its neighbours.
, meaning that the system may move to the new state even when it is worse (has a higher energy) than the current one. It is this feature that prevents the method from becoming stuck in a local minimum—a state that is worse than the global minimum, yet better than any of its neighbours.
On the other hand, when goes to zero, the probability must tend to zero if , and to a positive value if 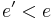 . That way, for sufficiently small values of , the system will increasingly favor moves that go "downhill" (to lower energy values), and avoid those that go "uphill". In particular, when becomes 0, the procedure will reduce to the greedy algorithm—which makes the move only if it goes downhill.
. That way, for sufficiently small values of , the system will increasingly favor moves that go "downhill" (to lower energy values), and avoid those that go "uphill". In particular, when becomes 0, the procedure will reduce to the greedy algorithm—which makes the move only if it goes downhill.
In the original description of SA, the probability was defined as 1 when — i.e., the procedure always moved downhill when it found a way to do so, irrespective of the temperature. Many descriptions and implementations of SA still take this condition as part of the method's definition. However, this condition is not essential for the method to work, and one may argue that it is both counterproductive and contrary to its principle.
The function is usually chosen so that the probability of accepting a move decreases when the difference  increases—that is, small uphill moves are more likely than large ones. However, this requirement is not strictly necessary, provided that the above requirements are met.
increases—that is, small uphill moves are more likely than large ones. However, this requirement is not strictly necessary, provided that the above requirements are met.
Given these properties, the evolution of the state depends crucially on the temperature . Roughly speaking, the evolution of is sensitive to coarser energy variations when is large, and to finer variations when is small.
The annealing schedule
Another essential feature of the SA method is that the temperature is gradually reduced as the simulation proceeds. Initially, is set to a high value (or infinity), and it is decreased at each step according to some annealing schedule—which may be specified by the user, but must end with 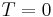 towards the end of the allotted time budget. In this way, the system is expected to wander initially towards a broad region of the search space containing good solutions, ignoring small features of the energy function; then drift towards low-energy regions that become narrower and narrower; and finally move downhill according to the steepest descent heuristic.
towards the end of the allotted time budget. In this way, the system is expected to wander initially towards a broad region of the search space containing good solutions, ignoring small features of the energy function; then drift towards low-energy regions that become narrower and narrower; and finally move downhill according to the steepest descent heuristic.

|

|
|
Example illustrating the effect of cooling schedule on the performance of simulated annealing. The problem is to rearrange the pixels of an image so as to minimize a certain potential energy function, which causes similar colours to attract at short range and repel at a slightly larger distance. The elementary moves swap two adjacent pixels. These images were obtained with a fast cooling schedule (left) and a slow cooling schedule (right), producing results similar to amorphous and crystalline solids, respectively. |
|
It can be shown[4] that for any given finite problem, the probability that the simulated annealing algorithm terminates with the global optimal solution approaches 1 as the annealing schedule is extended. This theoretical result, however, is not particularly helpful, since the time required to ensure a significant probability of success will usually exceed the time required for a complete search of the solution space.
Pseudocode
The following pseudocode implements the simulated annealing heuristic, as described above, starting from state s0 and continuing to a maximum of kmax steps or until a state with energy emax or less is found. The call neighbour(s) should generate a randomly chosen neighbour of a given state s; the call random() should return a random value in the range ![[0, 1]](/2010-wikipedia_en_wp1-0.8_orig_2010-12/I/ccfcd347d0bf65dc77afe01a3306a96b.png) . The annealing schedule is defined by the call temp(r), which should yield the temperature to use, given the fraction r of the time budget that has been expended so far.
. The annealing schedule is defined by the call temp(r), which should yield the temperature to use, given the fraction r of the time budget that has been expended so far.
s ← s0; e ← E(s) // Initial state, energy.
sbest ← s; ebest ← e // Initial "best" solution
k ← 0 // Energy evaluation count.
while k < kmax and e > emax // While time left & not good enough:
snew ← neighbour(s) // Pick some neighbour.
enew ← E(snew) // Compute its energy.
if enew < ebest then // Is this a new best?
sbest ← snew; ebest ← enew // Save 'new neighbour' to 'best found'.
if P(e, enew, temp(k/kmax)) > random() then // Should we move to it?
s ← snew; e ← enew // Yes, change state.
k ← k + 1 // One more evaluation done
return sbest // Return the best solution found.
Actually, the "pure" SA algorithm does not keep track of the best solution found so far: it does not use the variables sbest and ebest, it lacks the first if inside the loop, and, at the end, it returns the current state s instead of sbest. While saving the best state is a standard optimization, that can be used in any metaheuristic, it breaks the analogy with physical annealing — since a physical system can "store" a single state only.
In strict mathematical terms, saving the best state is not necessarily an improvement, since one may have to specify a smaller kmax in order to compensate for the higher cost per iteration. However, the step sbest ← snew happens only on a small fraction of the moves. Therefore, the optimization is usually worthwhile, even when state-copying is an expensive operation.
Selecting the parameters
In order to apply the SA method to a specific problem, one must specify the following parameters: the state space, the energy (goal) function E(), the candidate generator procedure neighbour(), the acceptance probability function P(), and the annealing schedule temp(). These choices can have a significant impact on the method's effectiveness. Unfortunately, there are no choices of these parameters that will be good for all problems, and there is no general way to find the best choices for a given problem. The following sections give some general guidelines.
Diameter of the search graph
Simulated annealing may be modeled as a random walk on a search graph, whose vertices are all possible states, and whose edges are the candidate moves. An essential requirement for the neighbour() function is that it must provide a sufficiently short path on this graph from the initial state to any state which may be the global optimum. (In other words, the diameter of the search graph must be small.) In the traveling salesman example above, for instance, the search space for 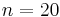 cities has
cities has 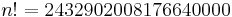 (2.4 quintillion) states; yet the neighbour generator function that swaps two consecutive cities can get from any state (tour) to any other state in maximum
(2.4 quintillion) states; yet the neighbour generator function that swaps two consecutive cities can get from any state (tour) to any other state in maximum 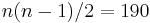 steps.
steps.
Transition probabilities
For each edge 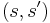 of the search graph, one defines a transition probability, which is the probability that the SA algorithm will move to state when its current state is . This probability depends on the current temperature as specified by temp(), by the order in which the candidate moves are generated by the neighbour() function, and by the acceptance probability function P(). (Note that the transition probability is not simply , because the candidates are tested serially.)
of the search graph, one defines a transition probability, which is the probability that the SA algorithm will move to state when its current state is . This probability depends on the current temperature as specified by temp(), by the order in which the candidate moves are generated by the neighbour() function, and by the acceptance probability function P(). (Note that the transition probability is not simply , because the candidates are tested serially.)
Acceptance probabilities
The specification of neighbour(), P(), and temp() is partially redundant. In practice, it's common to use the same acceptance function P() for many problems, and adjust the other two functions according to the specific problem.
In the formulation of the method by Kirkpatrick et al., the acceptance probability function was defined as 1 if , and 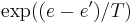 otherwise. This formula was superficially justified by analogy with the transitions of a physical system; it corresponds to the Metropolis-Hastings algorithm, in the case where the proposal distribution of Metropolis-Hastings is symmetric. However, this acceptance probability is often used for simulated annealing even when the neighbour() function, which is analogous to the proposal distribution in Metropolis-Hastings, is not symmetric, or not probabilistic at all. As a result, the transition probabilities of the simulated annealing algorithm do not correspond to the transitions of the analogous physical system, and the long-term distribution of states at a constant temperature need not bear any resemblance to the thermodynamic equilibrium distribution over states of that physical system, at any temperature. Nevertheless, most descriptions of SA assume the original acceptance function, which is probably hard-coded in many implementations of SA.
otherwise. This formula was superficially justified by analogy with the transitions of a physical system; it corresponds to the Metropolis-Hastings algorithm, in the case where the proposal distribution of Metropolis-Hastings is symmetric. However, this acceptance probability is often used for simulated annealing even when the neighbour() function, which is analogous to the proposal distribution in Metropolis-Hastings, is not symmetric, or not probabilistic at all. As a result, the transition probabilities of the simulated annealing algorithm do not correspond to the transitions of the analogous physical system, and the long-term distribution of states at a constant temperature need not bear any resemblance to the thermodynamic equilibrium distribution over states of that physical system, at any temperature. Nevertheless, most descriptions of SA assume the original acceptance function, which is probably hard-coded in many implementations of SA.
Efficient candidate generation
When choosing the candidate generator neighbour(), one must consider that after a few iterations of the SA algorithm, the current state is expected to have much lower energy than a random state. Therefore, as a general rule, one should skew the generator towards candidate moves where the energy of the destination state is likely to be similar to that of the current state. This heuristic (which is the main principle of the Metropolis-Hastings algorithm) tends to exclude "very good" candidate moves as well as "very bad" ones; however, the latter are usually much more common than the former, so the heuristic is generally quite effective.
In the traveling salesman problem above, for example, swapping two consecutive cities in a low-energy tour is expected to have a modest effect on its energy (length); whereas swapping two arbitrary cities is far more likely to increase its length than to decrease it. Thus, the consecutive-swap neighbour generator is expected to perform better than the arbitrary-swap one, even though the latter could provide a somewhat shorter path to the optimum (with 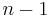 swaps, instead of
swaps, instead of 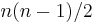 ).
).
A more precise statement of the heuristic is that one should try first candidate states for which 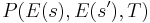 is large. For the "standard" acceptance function above, it means that
is large. For the "standard" acceptance function above, it means that 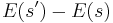 is on the order of or less. Thus, in the traveling salesman example above, one could use a neighbour() function that swaps two random cities, where the probability of choosing a city pair vanishes as their distance increases beyond .
is on the order of or less. Thus, in the traveling salesman example above, one could use a neighbour() function that swaps two random cities, where the probability of choosing a city pair vanishes as their distance increases beyond .
Barrier avoidance
When choosing the candidate generator neighbour() one must also try to reduce the number of "deep" local minima — states (or sets of connected states) that have much lower energy than all its neighbouring states. Such "closed catchment basins" of the energy function may trap the SA algorithm with high probability (roughly proportional to the number of states in the basin) and for a very long time (roughly exponential on the energy difference between the surrounding states and the bottom of the basin).
As a rule, it is impossible to design a candidate generator that will satisfy this goal and also prioritize candidates with similar energy. On the other hand, one can often vastly improve the efficiency of SA by relatively simple changes to the generator. In the traveling salesman problem, for instance, it is not hard to exhibit two tours  ,
,  , with nearly equal lengths, such that (0) is optimal, (1) every sequence of city-pair swaps that converts to goes through tours that are much longer than both, and (2) can be transformed into by flipping (reversing the order of) a set of consecutive cities. In this example, and lie in different "deep basins" if the generator performs only random pair-swaps; but they will be in the same basin if the generator performs random segment-flips.
, with nearly equal lengths, such that (0) is optimal, (1) every sequence of city-pair swaps that converts to goes through tours that are much longer than both, and (2) can be transformed into by flipping (reversing the order of) a set of consecutive cities. In this example, and lie in different "deep basins" if the generator performs only random pair-swaps; but they will be in the same basin if the generator performs random segment-flips.
Cooling schedule
The physical analogy that is used to justify SA assumes that the cooling rate is low enough for the probability distribution of the current state to be near thermodynamic equilibrium at all times. Unfortunately, the relaxation time—the time one must wait for the equilibrium to be restored after a change in temperature—strongly depends on the "topography" of the energy function and on the current temperature. In the SA algorithm, the relaxation time also depends on the candidate generator, in a very complicated way. Note that all these parameters are usually provided as black box functions to the SA algorithm.
Therefore, in practice the ideal cooling rate cannot be determined beforehand, and should be empirically adjusted for each problem. The variant of SA known as thermodynamic simulated annealing tries to avoid this problem by dispensing with the cooling schedule, and instead automatically adjusting the temperature at each step based on the energy difference between the two states, according to the laws of thermodynamics.
Restarts
Sometimes it is better to move back to a solution that was significantly better rather than always moving from the current state. This is called restarting. To do this we set s and e to sbest and ebest and perhaps restart the annealing schedule. The decision to restart could be based on a fixed number of steps, or based on the current energy being too high from the best energy so far.
Related methods
- Quantum annealing uses "quantum fluctuations" instead of thermal fluctuations to get through high but thin barriers in the target function.
- Stochastic tunneling attempts to overcome the increasing difficulty simulated annealing runs have in escaping from local minima as the temperature decreases, by 'tunneling' through barriers.
- Tabu search normally moves to neighbouring states of lower energy, but will take uphill moves when it finds itself stuck in a local minimum; and avoids cycles by keeping a "taboo list" of solutions already seen.
- Stochastic gradient descent runs many greedy searches from random initial locations.
- Genetic algorithms maintain a pool of solutions rather than just one. New candidate solutions are generated not only by "mutation" (as in SA), but also by "combination" of two solutions from the pool. Probabilistic criteria, similar to those used in SA, are used to select the candidates for mutation or combination, and for discarding excess solutions from the pool.
- Graduated optimization digressively "smooths" the target function while optimizing.
- Ant colony optimization (ACO) uses many ants (or agents) to traverse the solution space and find locally productive areas.
- The cross-entropy method (CE) generates candidates solutions via a parameterized probability distribution. The parameters are updated via cross-entropy minimization, so as to generate better samples in the next iteration.
- Harmony search mimics musicians in improvisation process where each musician plays a note for finding a best harmony all together.
- Stochastic optimization is an umbrella set of methods that includes simulated annealing and numerous other approaches.
- Particle swarm optimization is an algorithm modelled on swarm intelligence that finds a solution to an optimization problem in a search space, or model and predict social behavior in the presence of objectives.
- Intelligent Water Drops (IWD) which mimics the behavior of natural water drops to solve optimization problems
See also
- Adaptive simulated annealing
- Markov chain
- Combinatorial optimization
- Automatic label placement
- Multidisciplinary optimization
- Place and route
- Traveling salesman problem
- Reactive search
- Graph cuts in computer vision
- Particle swarm optimization
- Intelligent Water Drops
References
- ↑ Kirkpatrick, S.; C. D. Gelatt, M. P. Vecchi (1983-05-13). "Optimization by Simulated Annealing". Science. New Series 220 (4598): 671–680. doi:10.1126/science.220.4598.671. ISSN 00368075. PMID 17813860. http://www.jstor.org/stable/1690046. Retrieved 2009-01-16.
- ↑ V. Cerny, A thermodynamical approach to the travelling salesman problem: an efficient simulation algorithm. Journal of Optimization Theory and Applications, 45:41-51, 1985
- ↑ N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, and E. Teller. "Equations of State Calculations by Fast Computing Machines". Journal of Chemical Physics, 21(6):1087-1092, 1953. [1]
- ↑ Granville, V.; M. Krivanek, J.-P. Rasson (June 1994). "Simulated annealing: A proof of convergence". IEEE Transactions on Pattern Analysis and Machine Intelligence 16 (6): 652–656. doi:10.1109/34.295910.
- A. Das and B. K. Chakrabarti (Eds.), Quantum Annealing and Related Optimization Methods, Lecture Note in Physics, Vol. 679, Springer, Heidelberg (2005)
- E. Weinberger, Correlated and Uncorrelated Fitness Landscapes and How to Tell the Difference, Biological Cybernetics, 63, No. 5, 325-336 (1990).
- J. De Vicente, J. Lanchares, R. Hermida, "Placement by Thermodynamic Simulated Annealing”, Physics Letters A,Vol. 317, Issue 5-6, pp.415–423, 2003.
External links
- Simulated Annealing visualization A visualization of a simulated annealing solution to the N-Queens puzzle by Yuval Baror.
- Simulated Annealing A Java applet that allows you to experiment with simulated annealing. Source code included.
- "General Simulated Annealing Algorithm" An open-source MATLAB program for general simulated annealing exercises.
- Self-Guided Lesson on Simulated Annealing A Wikiversity project.
- Google in superposition of using, not using quantum computer Ars Technica discusses the possibility that the D-Wave computer being used by google may, in fact, be an efficient SA co-processor